February 26, 2019
Email Parser processes the emails running items (
Email sources,
Parsers and
Actions) one after another,
in a sequential way. In each step,
a set of fields are created based on the results of the previous steps. For instance, an
Email Source creates the field Subject and then a
Parser can use this field to capture, let’s say, an invoice number.
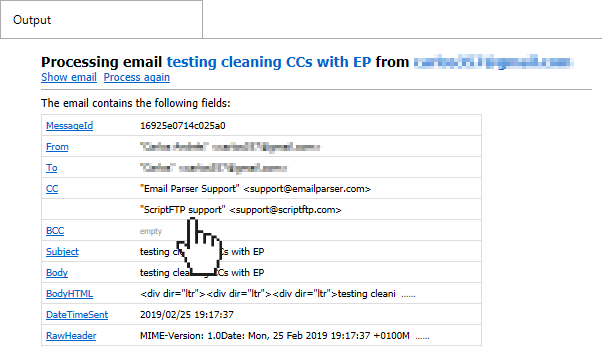
There are fields, like the To, CC and BCC that can contain multiple values. For example, the following email contains two values in the CC field:
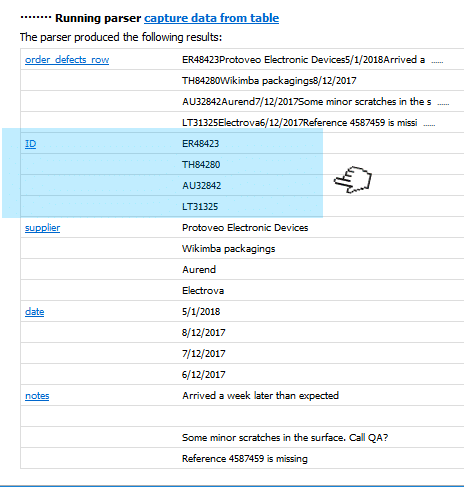
Multiple field values is something that also happens as a result of parsing, for instance, the body of an email:
Having the values separated, like in an spreadsheet, is what makes most sense in the majority of the cases. But it could happen that you need them merged into a single text value. In order to get this you have to run a small script like the following:
read more …
January 8, 2019
Email Parser automatically uses your local time (as set in Windows) to give a value to the fields DateTimeSent and DateTimeProcessed. But you may need to have these times adjusted to UTC+0 time or another time zone. For this you need to use a small script:
read more …
August 10, 2018
Recently, a user has posted an
interesting question in this website forum:
I’m writing to a text file.
Here’s my “Text to insert”:
‘1’,'<%customerID%>’,'<%customerID%>’,”,”,”,”,’ADMIN’,'<%shipToID%>’,”,”,”,”,N’
I’d like some way to increment the “1” with each successive line I extract from the e-mail….
I’ve been trying to create a static field above, and increment it.. tried a script & do it in C#…
Help!!!
and Thanks,
Rich
In this case, the user wanted an index value to
write to a text file but it may also happen that you need an index value to insert to a database table. If doing so note that database tables can
generate their own index when the
SQL INSERT command is sent. But you can also provide them your own index value manually if you want to.
For example, if we retrieve the following fields from an incoming email:
read more …
November 1, 2016
It is not a very common use case but an user has asked if it is possible to save an
attached file to a
database. We have written an example about that and posted to the examples section. You can find it
here.
October 3, 2016
Usually, when you want to parse a set of emails that meet a specific condition this condition is like “
emails contining the word ‘summary’ in the subject” or the more complex one”e
mails matching the regular expression ‘\d\d\d-\d\d\d'” in the body text“. But, what happens if you want the contrary? It is not so common, but sometimes you want to get only the emails that do not match a condition.
In these cases, the easiest way is to create an email filter that matches the condition, for example, emails contining the word ‘summary’ in the subject. And then, in the following steps of the program select as the source of emails the emails
not matching that filter. The screenshots below show how can be done:
read more …
August 11, 2016
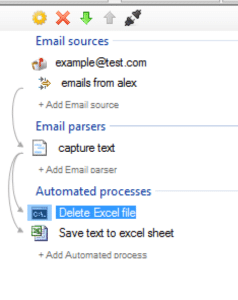
By default, the
process that inserts new rows in an Excel file keeps the existing rows from previous runs and adds the new captured data in a new row. If you want not to keep the previously captured data an easy way to accomplish that is to
create a new process that deletes the Excel file. A empty new Excel file will be automatically created the next time a row needs to be created. For example:

The command is:
del /f "C:\Users\Carlos\Desktop\invoices.xlsx"
June 12, 2014
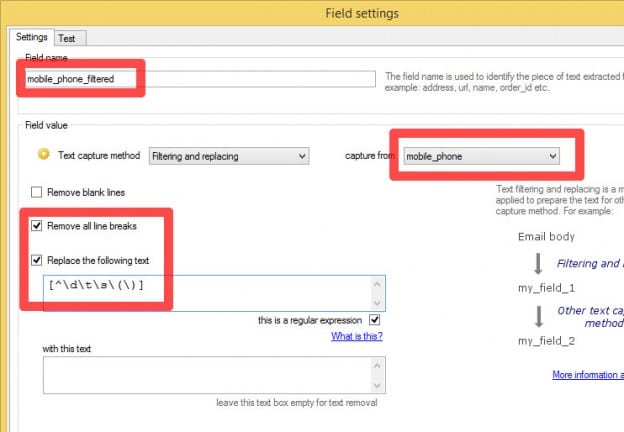
You can master regular expressions or c# scripting and build a parser that captures from the email exactly what you want but sometimes unwanted characters such as line breaks, tabs or weird characters (for instance ıġħť p̀ł) are part of the text captured.
To solve these issues we need to apply to the captured text another step of parsing called “text filtering and replacing” :
 read more …
read more …
May 22, 2014
Email Parser can save attached files from an incoming email to a given folder with the “Save attachments” action. Setting this up is quite easy and straightforward as it only requires to enter the folder where the attachments will be saved. But in the case you want to save the file contents to a database this requires a bit of more work. This blog post shows how it can be done in a Microsoft SQL server database.
read more …
May 16, 2014
CSV (Comma Separated Values) files are plain text files used to store tabular data, like Excel files or database tables, although in a much more limited way. Each line of the file stores a record (row) and each value (column) is separated by a comma character. Sometimes a tab, a semicolon or other character is used as separator.
For example, a CSV file contains the following data:
02/01/2013;22.43;1.1
02/02/2013;21.54;1.2
02/03/2013;19.43;1.0
02/04/2013;18.56;1.1
02/05/2013;19.21;1.1
02/06/2013;19.12;1.1
02/07/2013;20.56;1.0
02/08/2013;21.65;0.9
02/09/2013;22.54;1.0
02/10/2013;23.12;1.1
02/11/2013;23.87;1.0
02/12/2013;20.32;1.0
read more …
{kind=link}